How does insulin regulate the pathways for the conversion of body fat into ketone bodies? Why are ketone bodies overproduced in uncontrolled diabetes and what are the metabolic consequences.
1 Introduction
Production of ketone bodies (ketogenesis) occurs in the liver (and the kidneys) as an response to prevent hypoglycemia during starvation, and this process is normally suppressed and stimulated by insulin and glucagon, respectively. In addition to its physiological role in fasting, ketogenesis also has high activity in diabetes patients, which may lead to the potentially lethal condition called diabetic ketoacidosis.
2 Regulation of Ketogenesis by Insulin
Insulin suppresses ketogenesis by several mechanisms.
The steps of ketogenesis are breifly shown in Figure 2.1. There are three control points by which ketogenesis can be regulated by insulin: (1) adipocyte lipolysis, (2) mitochondrial fatty acid entry in ketogenic cells (mainly hepatocytes), (3) channeling of Acetyl-CoA (AcCoA) into the ketogenic pathway, and (4) mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase (HMG-CoA synthase).
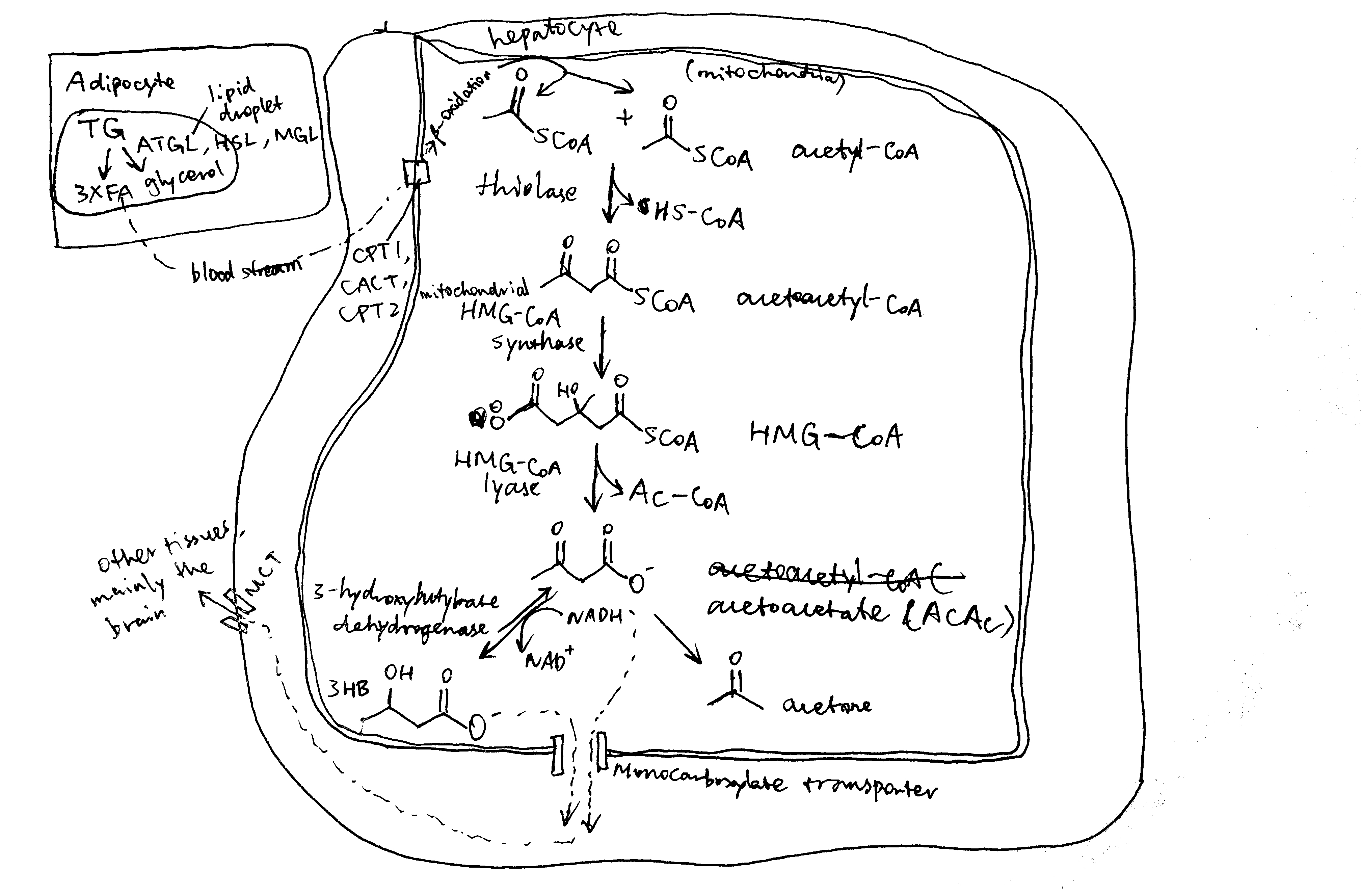
Figure 2.1: Production of ketone bodies from adipose-derived fatty acids.
2.1 Adipocyte Lipolysis
The major source molecules for ketogenesis are fatty acids obtained via lipolysis in white adipose tissues. Lipolysis involves the sequential hydrolysis of triacylglycerol (TAG) into 3 non-esterified fatty acids (NEFA) and glycerol by adipocyte triglyceride lipase (ATGL), hormone-sensitive lipase (HSL) and monoglyceride lipase (MGL). Activation of ATGL and HSL are dependent on PKA, which is activated by cAMP that are produced when catecholamines bind to cell-surface \(\beta\) adrenergic receptors.
Insulin is a potent inhibitor of lipolysis. It activates PKB via IRS and PI3K, and PKB in turn phosphorylates and activates phosphodiesterase 3B (PDE-3B). PDB-3B decyclises cAMP, thus diminishing PKA, and thus lipolytic, activity (Figure 2.2). During starvation, insulin levels are low, and adipocyte lipolysis is released from inhibition. Adipocytes are then able to release NEFA into bloodstream.
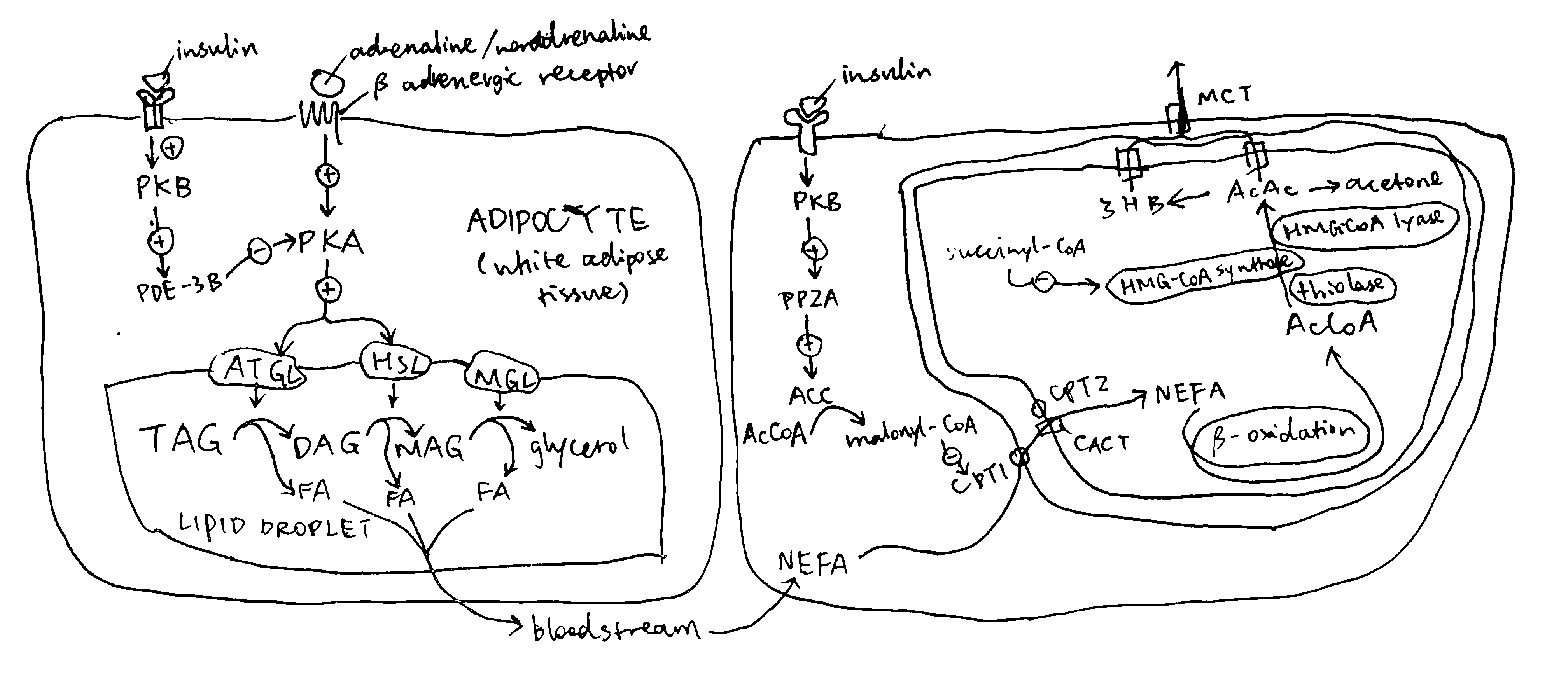
Figure 2.2: Left, regulation of lipolysis by insulin in adipocytes; right, regulation of uptake of long-chain fatty acids into mitochondria by insulin.
2.2 Regulation of CPT1
Once NEFA molecules arrive at hepatocytes, they are first broken down into AcCoA via \(\beta\)-oxidation before conversion into ketone bodies. Long chain fatty acids needs the carnitine shuttle to enter mitochondria where \(\beta\)-oxidation takes place, and thus the first enzyme of this shuttling system, carnitine palmitoyl transferase 1 (CPT1) is the rate limiting step and is under control. CPT1 is inhibited by malonyl CoA, which is the lipogenetic precursor produced by acetyl-CoA carboxylase (ACC). Insulin activates protein phosphotase 2A (PP2A, which is also activated by xylulose 5-phosphate during fed state), which dephosphorylates and activates ACC, thus promoting lipogenesis and suppressing \(\beta\)-oxidation and ketogenesis (Figure 2.2).
During starvation when insulin levels are low, the high fatty acid concentration in hepatocytes activates the transcription factor PPAR\(\alpha\) (activated by direct binding of fatty acids to PPAR\(\alpha\)), which in turn promotes transcription of several enzymes involved in lipid catabolism, including CPT1.
2.3 Channeling of Acetyl-CoA into Ketogenesis
AcCoA produced from NEFA via \(\beta\)-oxidation can either enter the Krebs cycle or the ketogenesis pathway. When the rate of production of AcCoA exceeds the capacity of citrate synthesis (when there is insufficient oxaloacetate), the surplus becomes the substrate for ketogenesis. This situation occurs during starvation, when insulin levels are low and glucagon levels are high, and gluconeogenesis active. Glucagon has several mechanisms to promote gluconeogenesis (and suppress glycolysis). For example, it activates PKA, which in turn activates the transcription factor CREB, which promotes transcription of gluconeogenenic enzymes such as PEP carboxykinase and glucose 6-phosphatase. Insulin, on the contary, suppresses gluconeogenesis. For instance, it activates PKB, which in turn phosphorylates and thus inactivates FOXO1, which is a transcription factor that promotes the transcription of a similar set of gluconeogenic enzymes. Insulin inhibits gluconeogenesis and thus ketogenesis.
2.4 Mitochondrial HMG-CoA Synthase
Mitochondrial HMG-CoA catalyses the irreversible step in ketogenesis, and therefore is a site of control.
Insulin promotes glycolysis and increases flux through the Krebs cycle. One of the Krebs cycle intermediate, succinyl-CoA, inactivates mitochondrial HMG-CoA by succinylation. Glucagon, on the contrary, activates gluconeogenesis, which causes depletion of Krebs cycle intermediates including succinyl-CoA, thus activating ketogenesis.
In addition, the transcription factor PPAR\(\alpha\) described previously also activates transcription of HMG-CoA synthase.
3 Diabetes and Ketone Bodies
3.1 Aetiology of Diabetes
Diabetes is characterised by insufficient insulin activity, which could be caused by either lack of insulin production or insulin resistance in target cells. Most diabetes cases fall into two categories, type 1 and type 2.
- Type 1 diabetes is caused by autoimmunity agaist pancreatic \(\beta\) cells, which leads to their death and inadequacy of insulin. The primary risk factor for type 1 diabetes is genetic, although several environmental risk factors, such as infection by viruses that show tropism to pancreatic islets.
- Type 2 diabetes begins with insulin resistance, but reduction of insulin production and number of \(\beta\) cells may also be implicated as the disease advances. Type 2 diabetes also has genetic risk factors, but environmental factors, such as sedentarity and obesity, are more important.
Whichever the type, ketogenesis is hyperactive in diabetes patients (although generally more severe in type 1) due to inadequate insulin activity.
3.2 Overproduction of Ketone Bodies
Given the fact that insulin suppresses ketogenesis and insulin activity is decreased in diabetic patients, it is easy to comprehend why ketone bodies are overproduced in diabetic individuals.
Lack of insulin activity upon adipocytes leads to uncontrolled lipolysis and subsequent release of non-esterified fatty acids. As fatty acids enter the liver, they are shuttled into mitochondria, broken down into acetyl-CoA via \(\beta\)-oxidation and converted to ketone bodies. This process is facilitated by the low insulin activity, which activates gluconeogenesis (e.g. via FOXO1-induced transcription of gluconeogenenic enzymes and phosphorylation of PFK2/FBPase2 bifunctional enzyme), which in turn releases the rate-limiting enzyme, HMG-CoA from succinylation inhibition and reduces usage of acetyl-CoA in Krebs cycle. The low insulin activity also inhibits ACC, thus activating CPT1 and hence \(\beta\)-oxidation. The high fatty acid concentration also leads to activation of PPAR\(\alpha\), which promotes transcription of ketogenic enzymes such as CPT-1 and HMG-CoA synthatase.
3.3 Consequences of Ketoacidosis
Diabetic ketoacidosis (DKA) the most common acute emergency in diabetes patients. It is a potentially lethal condition that is characterised by lowered blood pH due to accumulation of acidic ketone bodies (acetoacetate and 3-hydroxybutyrate). DKA is caused by lack of insulin and thus occurs mostly in uncontrolled type 1 diabetes (DKA is not commonly seen in type 2 diabetes, because even a tiny amount of insulin activity would inhibit ketogenesis; however, there is a small proportion of “ketosis-prone type 2 diabetes”). Often, DKA is triggered by stress or illness, which are associated with increase of ketogenesis-promoting hormones, glucagon and catecholamines.
Ketoacidosis has several consequences, as described below.
- Kussmaul breathing: initally, bicarbonate ions buffur protons dissociated from ketone bodies, but this buffering system quickly becomes overwhelmed. To compensate, blood CO2 concentration is reduced by deep, regular breaths known as Kussmaul breathing.
- Hypovolaemia: hyperglycemia and ketonaemia results in excretion of glucose and ketone bodies (which normally should be absorbed) in urine. Since they are osmotically active, their presence in urine causes osmotic diuresis, which further leads to hypovolaemia (decrease in body fluid volume).
- Electrolyte disturvance: as the the plasma pH falls, extracellular protons are exchanged for intracellular potassium ions. In addition, owing to hypovolaemia, the aldosterone concentration increases to retain water, but this hormone also leads to excretion of K+ and retention of Na+.
- The low plasma pH impair with many biological functions, especially in the brain, where potentially life-threatening coma and cerebral oedema may occur.