Title
You want to over-express a protein secreted by the malarial parasite. It is cysteine-rich and has no known structure. Outline an expression and purification strategy, using both traditional and high-throughput methods, to produce a sample suitable for protein crystallography. Include a description of how you would assess the quality of the purified protein.
1 Overview
The general workflow for obtaining a sample of (malarial) protein is summarised as follows:
- Identifying and amplifying the gene of interest (GOI)
- Constructing the vector
- Transformation/transfection of host cells
- Screening for the most viable (high yield and solubility) transformants
- Extracting and purifying proteins
For crystallography studies, a protein sample with high purity, solubility and yield must be obtained. The expression and purification strategies depend on the characteristics of the protein of interest, such as size, shape, intrinsic solubility, requirement of post-translational modifications and/or cofactors, presence of disulfide bridges, in vitro stability.
2 Protein Expression
2.1 Identifying and amplifying the gene
If the coding sequence of the protein is unknown but the protein is purified, mass spectrometry (or chemical methods such as Edman degradation) can be used to determine the amino acid sequence of the protein, which can then be used to search for the the corresponding DNA sequence.
Usually, instead of using the full DNA sequence, cDNA is used in cloning because it does not contain introns. cDNA is made by:
- extracting total RNA from cells by TRIzol and then isolate the mature mRNA by affinity chromatography (with poly-T coated resins which binds to poly-A tails of mature mRNA)
- reverse transcription of the template DNA strand by viral reverse transcriptase (RT) followed by RNA degradation (often by the RNAse H activity of RT)
- synthesis of the coding strand by DNA polymerase
Often, the latter two steps are done repeatedly in RT-PCR to amplify the DNA fragment, if the sequence of the GOI is known and specific1 primers are designed. The primers include additional restriction enzyme cleavage sites for the convinience of traditional ligation-dependent cloning, or other sequences for ligation-independent cloning (see Section 2.3).
If the protein is already known to have a match in malarial cDNA library, we can skip most of the above steps and PCR-amplify the cDNA of interest from the library directly.
When we know the sequence of GOI, we can do a BLAST to find possible homologous proteins, and study any relavant scientific literature. The structure of some homologous proteins might have been solved previously and we can refer to their expression and purification strategies, which might prevent some waste of time on trial-and-errors. Bioinformatic analyses, such as disorder prediction using the RONN algorithm and generation of 3D homology models using the Phyre2 server, might also be helpful.
Codon optimisation might also be needed. Codon usage can vary significantly between species as well as between genome types such as nuclear DNA and mitochondrial DNA. For example, the frequencies of codons AGG, AGA, and CGA (which code for arginine) in H. sapiens are 11.4, 11.5 and 6.3, respectively, while the corresponding frequencies in E. coli are 1.2, 2.1 and 3.6. Codons that occur in high frequency in Plasmodium but in low frequencies in E. coli would result in a condition where the pool of tRNA for that codon will be so low as to become depleted. When the rare tRNAs are depleted to produce the recombinant protein, proliferation of the host cells is restricted, leading to low yield. This problem may be partially solved by the use of an inducible expression vector (e.g. IPTG), but when the foreign gene contains a large number of rare codons, this is not enough. There are two further approaches to improve the yield. First, the rare codons in the foreign genes can be substituted with prevalent codons, but this is not always reliable. Second, host cells can be transformed with the genes that code for the rare tRNAs. This approach is reliable and several such cell lines are commercially available. Custom host cells can also be made using plasmids.
We might fail in crystallising the single full-length protein (often because of low solubility). In that case, we can try co-expressing another protein or add an appropriate cofactor and crystallising the resulting complex. If this still fails, we might consider truncating the GOI, e.g. removing a very hydrophobic region that results in low solubility. In high-throughput approaches, such as the pipeline adopted by Oxford Protein Production Facility (OPPF), many variants are constructed in parallel to maximise output.
2.2 Choosing the host organism and the vector
2.2.1 Bacteria and plasmid vectors
Typically and traditionally, the gene of interest (GOI) is incorporated into a plasmid vector via a ligation-dependent mechanism, then the plasmid is introduced into bacterial (E. coli) cells for expression. There are also ligation-independent methods for making vectors, which is more robust and easy to use (only need to design one ‘adaptor’ parts of the primers once), making it suitable for high-throughput methods.
Bacteria-based protein expression is time- and cost-efficient, but it has two major limitations:
- the insertion size of a plasmid is small, typically 2-10 kb (bacterial artificial chromosomes (BAC) with greater insertion sizes can be used, but the procedures are more complex)
- Bacteria might be unable provide the appropriate protein folding environment (including chaperones) and/or post-translational modifications that can be crucial to protein’s functions.
Given the protein of interest is cysteine-rich, it is susceptible that disulfide bridges are present if this protein is also extracellular. The normal intracellular reducing environment disfavours disulfide bridge formation, but Origami and related strains have an oxidising intracellular environment due to mutant thioredoxin reductase and glutathione reductase, and they are used to express proteins that require disulfide bonds to achieve their correctly-folded conformation.
The plasmid should contain selectable markers for easy identification of successful transformants. The combination of AmpR and lacZ\(\alpha\) is a common choice, as explained in Figure 2.1.
In addition, the promoter should be strong and tightly regulated with minimum or complete lack of basal level transcription under uninduced conditions. The IPTG inducible T7 promoter system (as in pET plasmid) is one of the common choices. IPTG is an analog of allolactose, which can bind to and silence lac inhibitor, de-inhibiting the lac operator. IPTG is not digested by the bacteria, and thus its constant concentration continuously induces protein expression. The lac operator may be also made symmetric, strictly preventing transcription when uninduced.
Some commerially available plamids also include tags added to the N- or C-terminus of the insertion site (multiple cloning site), which aids protein purification. This can be a polyhistidine tag, a fusion protein (e.g. GST) or a short recognisable peptide, and often a cleavage sequence is also introduced for removal of the tags.
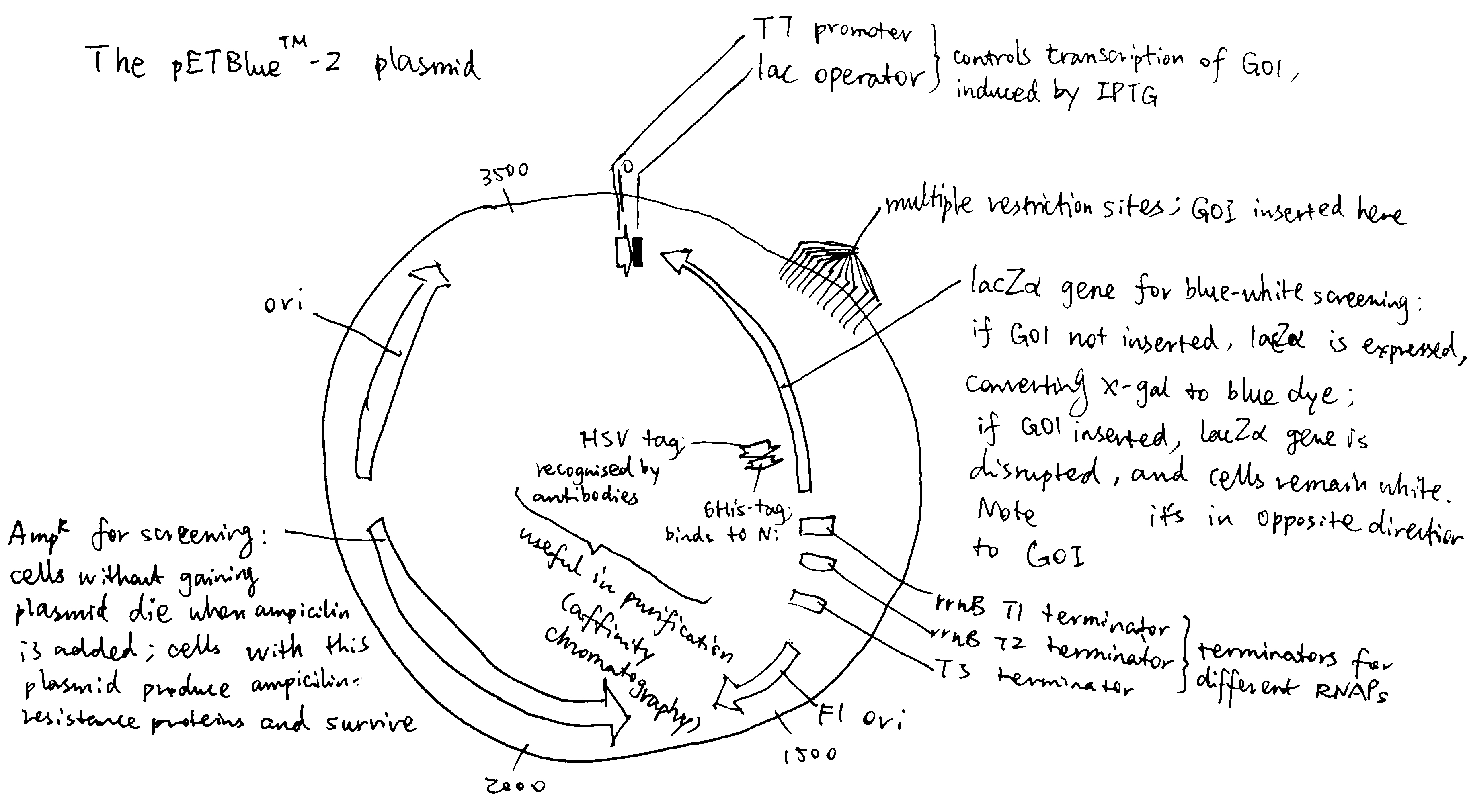
Figure 2.1: The pETBlue-2 plasmid vector
Plasmid-based bacterial transformation is usually permanent because plasmids can replicate themselves and be distributed into daughter cells after cell division.
2.2.2 Eukaryotic hosts
If the protein is incompatible for expression in bacteria, an eukaryotic host is chosen. This is typically yeast, insect, or human cells.
Transfection of eukaryotic cells can be transient or permanent. Eukaryotic cells do not transcribe plasmids, and the DNA fragment of interest must integrates into the host cell’s genome to produce a stable cell line–which is a rare event and takes more time to achieve. If we had to use a eukaryotic host, transient transfection may be used first to screen for the most viable construct, then a stable cell line is made with this construct for higher yield and convience for later use.
2.2.3 High-throughput strategy
High-throughput protein production aims to use robots to automate the steps in making expression constructs and screening them.
Take the OPPF pipeline for example, when the GOI is an eukaryotic protein, they test expressions with different vector constructs (with varying amino acid start and end points, fusion partners, and plasmids) in both E. coli and insect cells in parallel (Figure 2.2). The expresison screening is automated and performed in 96-well format.
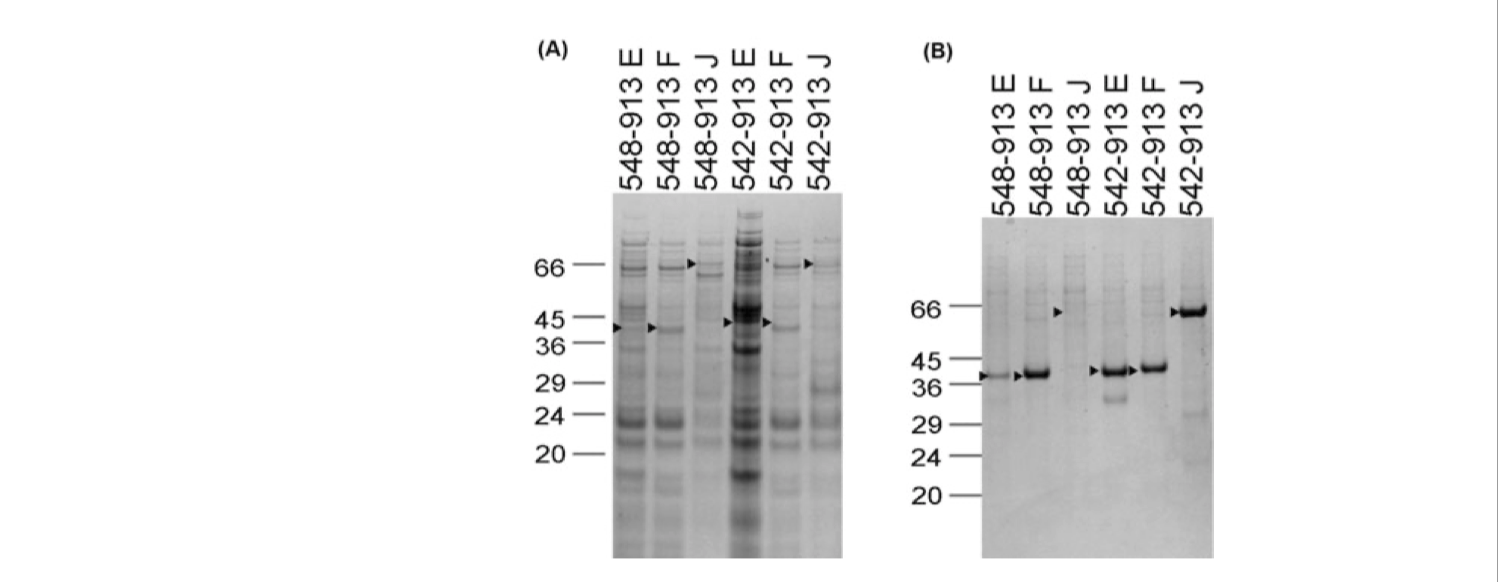
Figure 2.2: SDS-PAGE gel after NiNTA magnetic bead purification showing expression of constructs for DDR1 in (A) E. coli and (B) insect cells via the baculovirus system. The names of constructs correspond to amino acid start and end points and the vector being used (pOPINE, pOPINF or pOPINJ, which confers a C-terminal His-tag, an N-terminal His-tag, or an N-terminal His-GST tag, respectively)
they also used ligation-independent cloning methods (Section 2.3) to reduce variability and thus simplify the workflow.
2.3 Constructing the vector
The DNA fragment we obtained should be incorporated into a vector before they can be introduced into the host cell. Usually, a recombinant plasmid is made first and amplified in bacteria. Then, if necessary, it can be transferred into other vectors such as viruses and liposomes, depending on the strategies. Here I describe two ways of making recombinant plasmids: traditional restriction enzyme and ligation-based plasmid cloning, and T4 polymerase-based ligation-independent cloning (LIC).
Figure 2.3: Left, traditional cloning based on restriction enzyme and ligase; right, ligase-independent cloning based on T4 polymerase
As shown in Figure 2.3, in the ligation-dependent method, the PCR product contains flanking restriction sites (came from the designed primers) to be recognised by a restriction enzyme (e.g. EcoRI). The plasmid also contain the restriction site for EcoRI (located in the multiple cloning site, see Figure 2.1). Cleavage by EcoRI results in complementary ‘sticky ends’, which facilitate annealing of GOI to the plasmid. The sticky sequences are shart (usually 3-5 bp), thus, to overcome low stability, ligation must be done immediately.
The ligation-based cloning are not proper for high throughput protein production (HTPP) projects due to several disadvantages:
- incomplete DNA digestion and poor ligation yields
- each GOI has to be inspected for any internal restriction sites (for exclusion from the choices of the ligation sequence)
- unwanted amino acids can be introduced to the expressed protein
- the GOI can only be cloned in the vector position where the selected restriction site is present
Ligation-independent cloning (LIC) overcomes many of the problems described above. In a T4 DNA polymerase-dependent approach, the sticky sequences are made by the 3’-to-5’ exonuclease activity of T4 polymerase. These sequences are long, allowing formation of stable recombinant plasmid without in vitro ligase treatment. The nicks in sugar-phosphate backbone are later fixed by host ligase.
The obvious elegance of T4-LIC is that once the sticky sequence is designed and constructed, it can be used for any GOI. As T4 polymerase (exonuclease activity) always proceeds from 3’ ends, any internal sticky sequences will not be disrupting.
2.4 Transformation/Transfection
Transformation is the process of delivering GOI-containing foreign DNA into host cells. For eukaryotic cells, this is more often known as transfection (because ‘transformtion’ has other meanings). Many chemical/physical transformation/transfection methods are generally done by making transient holes on host cell plasma membrane (technically, ‘inducing competence’ in host cells). Here are two examples.
Heat-shock transformation is used for small vectors such as plasmids. The general procedures are:
- Host cells are incubated in a solution containing divalent cations (typically CaCl2) on ice.
- CaCl2 partially disrupts the cell membrane, which allows the recombinant DNA enter the host cell. Such cells are called competent cells.
- Cells are exposed to a heat pulse (heat shock), and the thermal imbalance causes the entry of DNA through disrupted plasma membrane.
Electroporation can be used for larger vectors such as BAC and PAC2. The general procedures are:
- Host cells are placed into a cuvette, together with the vector. The cuvette is connect to electrodes.
- The cuvette containing the mixture is subjected to intense electric pulses (2500 V/cm for bacteria, lower for animal and plant cells) each lasting for only a few milliseconds
- Most cells would die under such treatment, but for those survived, their membranes are polarised by the electric field and are disrupted, and DNA enters through the pores. Finally, the membrane reseals after the treatment.
Depending on the identity of the host cell and the vector, other methods are also available. These are not directly relevant with our goal of expressing a malarial protein, so here is a simple listing:
- cosmid and fosmid vectors are introduced into bacterial cells via bacteriophages
- calcium phosphate co-precipitation for mammalian cells
- microinjection for animal cells
- microprojectile bombardment for plant cells
- adenovirus and lentivirus vectors for mammalian cells
- Agrobacterium-mediated transformation for plant cells
- genome editing techniques based on ‘designer nucleases’ such as ZFN, TALEN and CRISPR-Cas9 with homology directed repair
3 Protein Extraction and Purification
After proliferation of transformed/transfected cells, we can extract and purify the protein fo interest.
3.1 Extraction
3.1.1 Cell lysis
The first step is to lyse, or homogenise the cells to release the protein. To do this, cells can be subjected to osmotic shock or ultrasonic vibration, forced through a small orifice, or ground up in a blender. (if a membrane protein is to be extracted, detergents are normally used, although detergent-free methods have been developed recently)
3.1.2 Removing cellular components and debris
Repeated centrifugation, each time with a higher speed (i.e. differential centrifugation), removes many impurities with large sizes. Organelles (and small vesicles and microsomes) sediment (forming pallets) while proteins remain in the solution (as supernatant).
A finer degree of separation can be achieved by layering the homogenate in a thin band on top of a salt solution that fills a centrifuge tube. When centrifuged, the various components in the mixture move as a series of distinct bands through the solution, each at a different rate, in a process called velocity sedimentation. A glucose gradient is established to protect the bands from convective mixing.
3.2 Purification
Classical methods for separating proteins depend on variable protein properties, including solubility, size, charge, and binding affinity. In most cases, protein mixtures are sequentially subjected to different separation methods, each based on a different property. After each step of purification, the fractions are assayed (see Section 3.2.3).
3.2.1 Salting out and dialysis
The addition of certain salts in the right amount can selectively precipitate the protein of interest, while others remaining in solution. The precipitation is removed by centrifugation.
Dialysis is then used to remove the salt from the solution containing the protein of interest. The protein mixture is place inside a dialysis bag (i.e. made of size-selective permeable membrane) and placed in a buffer solution with low salt concentration. Salt in the protein-containing solution then diffuse out, leaving proteins inside the bag.
3.2.2 Chromatography
In column chromatography, the solution containing proteins is passed through a column containing a solid matrix (resin). Different proteins are retarded to different extents by their interaction with the matrix, and they can be collected separately as they flow out of the bottom of the column. Depending on the choice of matrix, proteins can be separated according to their charge (ion-exchange chromatography),nhydrophobicity (hydrophobic chromatography), size (gel-filtration/size-exclusion chromatography), or ability to bind to particular small molecules or to other macromolecules (affinity chromatography). The last two are the most important for high throughput protein purification are affinity chromatography and size-exclusion chromatography.
Affinity chromatography extract protein according to the tags we attached to target proteins. For example, His-tagged proteins bind to Nickle-coated resin and HSV epitope tagged proteins bind to resin coated with corresponding antibodies (polyHis-Ni interactions often have lower specificity compared to enzyme-substrate and epitope-antibody interactions)
Size exclusion chromatography cleans abnormal protein aggregates based on their large size.
A problem experienced by column chromatographic methods is diffusional spreading (i.e. proteins that are going down faster also diffuse upwards, mixing with proteins going slower), which reduces the resolution. The degree of diffusional spreading increases with time during which proteins stay in the column. HPLC (high-performance liquid chromatography) solves this problem. It makes use of high-pressure pumps that speed the movement down the column, so that the time of travelling, and hence the diffusional spreading, is greatly reduced, leading to higher resolution.
3.2.3 Monitoring the progress of purification
As purification progresses:
- the total amount of protein should decrease as unwanted proteins are removed and some target proteins are lost
- amount of target proteins should also decrease because they are unavoidably lost e.g. by nonspecific attachment to the purification apparatus or washed away
- the proportion of the target protein (i.e. purity) should increase as unwanted proteins are removed
The simplest way to monitor these changes is running a SDS-PAGE gel electrophoresis after each purification step. The bands should progressively decrease to one (which correspond to the target protein), and the thickness of this band should increase because the concentration of target protein increases (its absolute amount decreases, but as we removed the fractions without the target protein in chromatography, its concentration should increase).
To assess whether the protein is correctly folded and functional, specific assays are used For example, if the target protein is an enzyme, it can be assayed using its substrate; if it is a protein which binds to another molecule, their interaction can be probed by surface plasmon resonance. Circular dichroism also helps to assess the foldedness of the target protein.
3.3 Preparation for crystallography
For crystallisation, the protein sample must be homogenous and correctly folded. They can be checked by the methods descibed in Section 3.2.3.
If the affinity tag needs to be removed (which is often required for structual studies), a protease cleavage site is often incorporated before or after the fusion tag and the cleavage can be conducted either in solution following purification (the protease themselves are tagged) or immediately after enzyme capture in situ on the chromatography resin itself. AKTA Express (GE Healthcare) is an elegant procedure for on-column cleavage coupled to multidimensional chromatography that is highly amenable to high throughput protein purification.
There are three methods to grow protein crystalls, namely ‘hanging drop’, ‘sitting drop’, and microdialysis, but the underlying principles are similar. The protein is first dissolved in a ‘crystallisation cocktail’ droplet and concentrated to 2-50 mg/ml, then it is allowed to equilibrate with a more concentrated reservoir solution of the cocktail with a volume ratio of 1:1. As the protein droplet becomes supersaturated, it may start to crystalise. Robots are commonly used for automatic screeening and optimisation of crystallisation conditions. If crystallisation fails under all conditions, we can try co-crystalising with a ligand. If this still fails, we might consider using a truncated protein, as described in Section 2.1.
References
Doyle, Sharon A. 2008. HIgh Throughput Protein Expression and Purification: Methods and Protocols. Edited by John W. Walker. Methods in Molecular Biology 498. Humana Press.
ThermoFisher. 2019. https://www.thermofisher.com/uk/en/home/references/gibco-cell-culture-basics/transfection-basics/gene-delivery-technologies.html.
Vicentelli, Renaud. 2019. HIgh-Throughput Protein Production and Purification: Methods and Protocols. Edited by John M. Walker. Vol. 2025. Methods in Molecular Biology. New York, NY: Humana Press.