1 Introduction
Many proteins that perform vital functions are membrane proteins. These include transport proteins (channels, transporters and pumps), cell adhesion molecules, and proteins that transduces energy in the electron transport chain. Despite their diverse roles, the same set of biophysical and biochemical rules govern their structural stability. Membrane proteins fall into two broad categories: 1) integeral proteins, which are embedded within a membrane and can only be isolated with detergents, and 2) peripheral proteins, which are associated with the surface of a membrane and can be removed without detergents. The focus of this essay is on how integral proteins interact with the membrane to maintain their structural stability.
2 The Biomembrane Environment
The basic structure of a biomembrane is a bilayer of amphipathic phospholipids and sphingolipids, where the polar head groups of lipids align the surfaces (each ~10 angstroms thick) and the acyl chains of lipid tails occupy the hydrophobic core (~ 30 angstroms thick). A bitopic or polytopic integral membrane protein spans the membrane entirely one or more times (Figure 2.1).
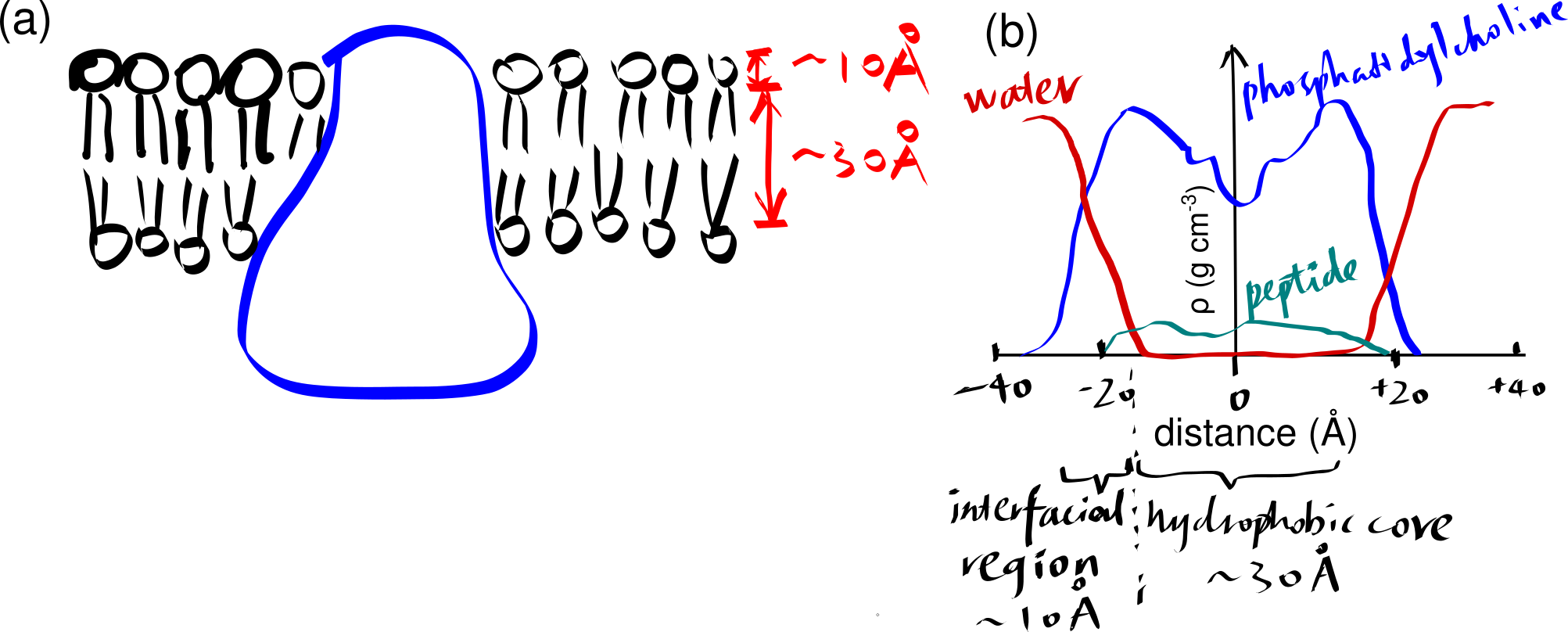
Figure 2.1: (a) a schematic showing a integral protein embedded within a biomembrane. (b)
The hydrophobic core region has a dielectric constant (\(\epsilon_{r}\)) of about 2, which is much lower than \(\epsilon_{r}\) of water (about 80), which means the same pair of charges separated by the same distance experience more electrostatic force in the membrane core than in water, according to the Coulomb’s law:
\[F = \dfrac{q_1 q_2}{4\pi \epsilon_0 \epsilon_r r^2}\]
This has several implications on acidic and basic amino acid residues. First, the carboxyl group of acidic side chains are more difficult to dissociate (i.e. p\(K_a\)s are shifted up), meaning they tend to remain in the uncharged (–COOH) form. Second, in \(\alpha\)-helical bundles, oppositely charged residues in adjacent helices associated more strongly than they do in aqueous environment. Third, this facilitates the snorkelling of lysine and arginine residues near the interface (Section 5).
3 The Thermodynamic Basis of Secondary Structure Formation
Transmembrane proteins tend to adopt a conformation that minimises the contact between their polar groups (main chain -NH, C=O, and side chains of polar residues, if any) and the hydrophobic core. This can be explained by simple thermodynamic reasoning. The water molecules in the aqueous environment outside the membrane are capable of forming relatively strong dipole-dipole interactions and hydrogen bonds (H-bonds) with polar (including charged) amino acids, while the non-polar alkyl groups that occupy the hydrophobic core of the membrane can only provide weak van der Waals interactions. Thus, if a protein adopts a conformation that exposes many polar groups, the loss of strong interactions with water will make this process very energetically unfavourable (very large \(\Delta G\)). On the contrary, if a protein with a hydrophobic surface were present in an aqueous environment, it would disrupt the dipole-dipole interactions and hydrogen bonding among water and other polar molecules. Thus, its insertion into the membrane is energetically favourable.
By forming either an \(\alpha\)-helix or a \(\beta\)-sheet (Figure 3.1), the hydrogen bonds between main chain -NH and -C=O are maximised, and this is an efficient way to shield the polar groups on the main chain from the hydrophobic core. Many transmembrane proteins, such as bacteriorhodopsin, contain multiple TM helices, which aggregates to form a \(\alpha\)-helix bundle. A \(\beta\)-sheet has to twist and coil to form a ring, called a \(\beta\)-barrel, in order to shield the main chain -NH and C=O groups on the first and last strands. \(\beta\)-barrels usually have a even number of strands so that every strand is anti-parallel to adjacent strands, there are exceptions: the voltage-dependent anion channels (VDACs) located on mitochondrial outer membrane has 19 strands, where the first strand is parallel with tha last strand, with weaker hydrogen bonding. \(\beta\)-barrels are less common than \(\alpha\)-helix bundles, and they are predominantly present on the outer membrane of Gram-negative bacteria, mitochondria, and chloroplasts.
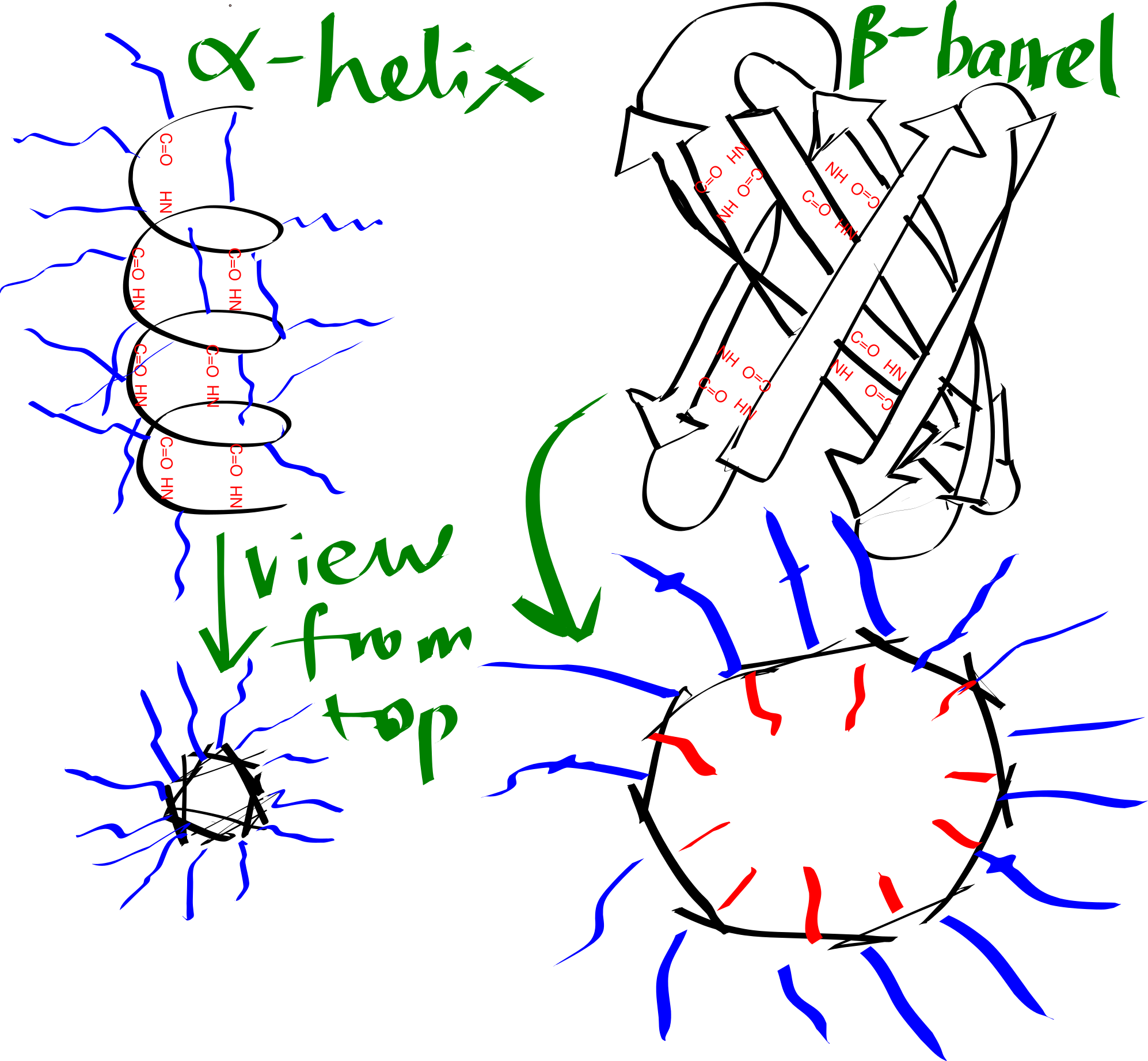
Figure 3.1: Schematic representation of an alpha helix and a beta barrel. Blue and red lines represent hydrophobic and hydrophilic side chains, respectively.
The transmembrane region of each helix in a \(\alpha\)-helical bundle is composed predominantly of hydrophobic amino acid residues (Ala, Leu, Ile, Val, Phe), which are exposed on the surface and allows favourable interaction with the hydrophobic core. By contrast, each \(\beta\)-strand of a \(\beta\)-barrel has an alternating pattern of hydrophobic and polar residues. This is because two adjacent residues in a beta strand have their side chains pointing in opposite directions, and a \(\beta\)-barrel formed in this way has an hydrophobic exterior that interacts with the hydrophobic core and an hydrophilic inner surface that is in contact with the aqueous environment.
Thus, TM helices, but not \(\beta\)-barrels, can be predicted using a hydropathy plot (Figure 3.2), in which the average hydrophobicity index of a fixed number of consecutive residues (a “window”), \(H(i)\), is plotted against the index (\(i\)) of the window, i.e.
\[H(i) = \sum_{i<j<i+k}h(a_j)/k\]
for \(1 < i < n - k\), where n is the length (number of residues) of the peptide, \(h(a_j)\) is the hydrophobicity index of the \(j\)-th amino acid residue and \(k\) is the window size.
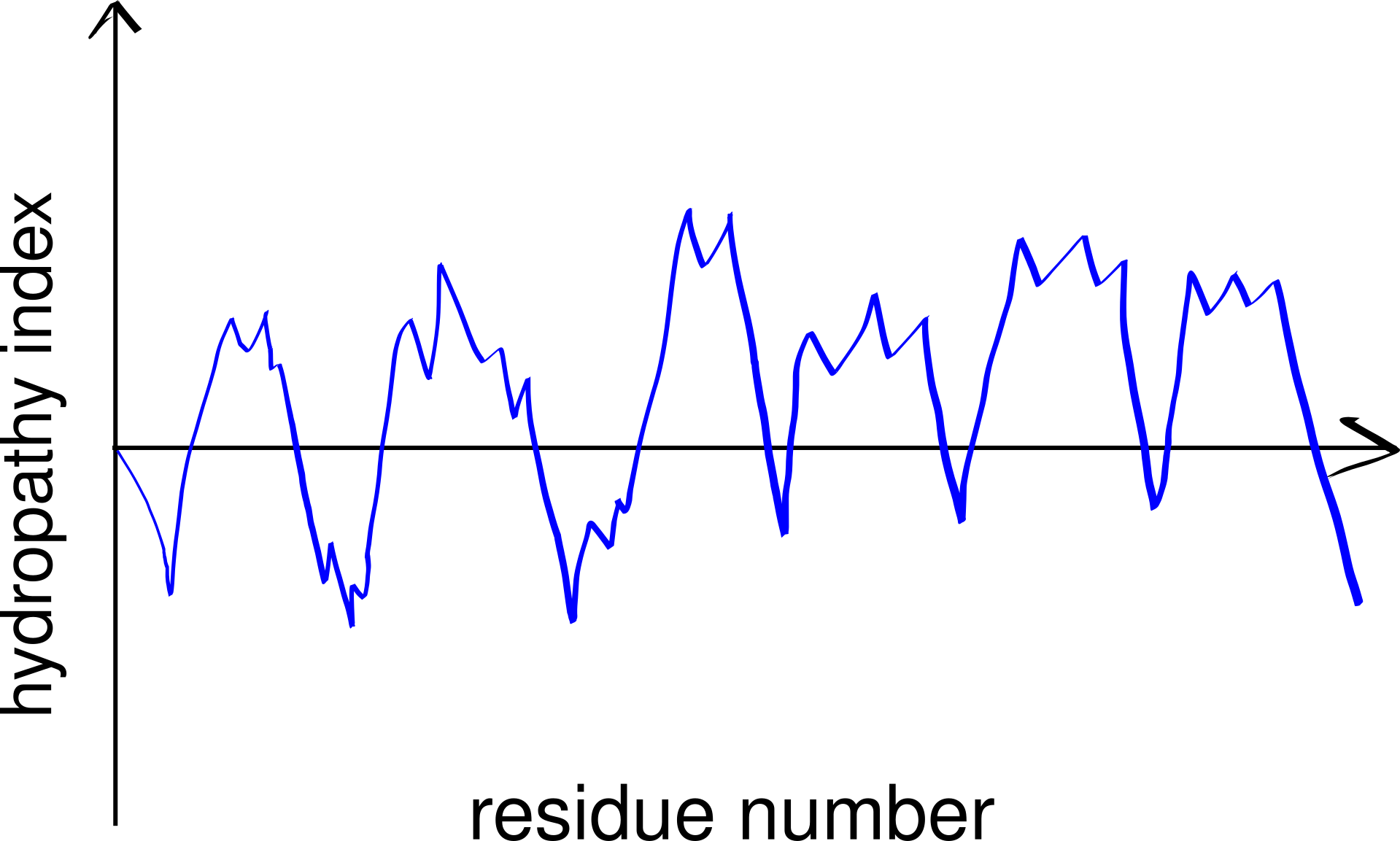
Figure 3.2: A schematic hydropathy plot. Every peak in the plot represents a highly hydrophobic local region in the peptide and thus indicates a potential transmembrane helix.
A small number of polar (even charged) residues within a TM helix can be tolerated as long as the overall transmembrane segment is hydrophobic enough. In addition, in polytopic proteins, polar residues in adjacent helices may help to stabilise each other. These non-hydrophobic residues often have functional roles, as exemplified by voltage-gated K+, Na+ and Ca2+ channels (Figure 3.3).
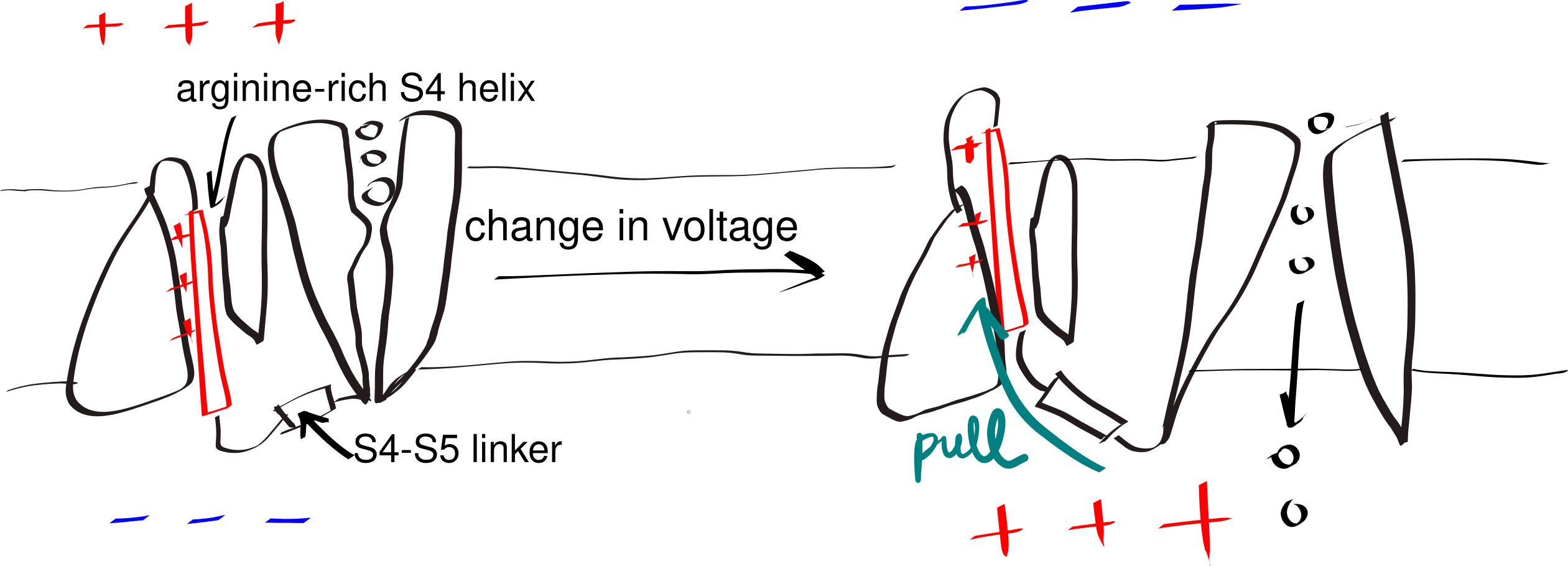
Figure 3.3: The gating mechanism of voltage-gated potassium channel. The S4 helix (red) is arginine-rich and thus positively charged. A linker connects S4 and S5 helix, the latter being a part of the pore domain. In resting state, the interior (bottom) of the cell is negatively charged relative to the exterior (top). A change in voltage causes the S4 helix to move upwards due to electrostatic force, and it pulls the gate open via the S4-S5 linker.
4 Glycine and Proline in TM Helices
The side chain of proline forms a pentameric ring with the amine group on the main chain. Thus, in a helix, the amine group of a proline does not have a hydrogen that can be H-bonded to the main chain C=O of the residue above it. This introduces local flexibility within a relatively regid helix and often forms a hinge. Similarly, due to glycine’s small size, it can tolerate a much wider range of dihedral angles than other amino acids, which also makes it able to introduce flexibility. This flexibility is crucial for the function of some proteins, such as voltage-gated ion channels (Figure 3.3).
5 Tryptophan and Tyrosine at the Interface and Lysine/Arginine Snorkelling
Tryptophan and tyrosine are often found in the interfacial region in both \(\alpha\)-helical bundle and \(\beta\)-barrel proteins. Their hydrophobic aromatic ring interacts with the hydrophobic core, and their polar -NH (of Trp) or -OH (of Tyr) groups interacts with the lipid head groups. Similarly, lysine and arginine extend their side chain towards the surface, so that their positively charged guanidium (of Arg) or amide (of Lys) groups form strong ionic interactions with the negatively charged lipid head groups. This phenomenon is known as “snorkelling.” These interactions helps to lock the membrane in place within the membrane, preventing vertical motions.
6 Interactions with Lipids
Membrane proteins interacts with lipid in various ways, and these interactions contribute to the structural stability and function of the protein to different extents.
A transmembrane protein interacts strongly with the shell (annulus) of lipid that surrounds it. These lipids are called annular lipids and can be distinguished experimentally from the bulk lipids of the bilayer. In addition, there is a third class of lipids, called nonannular lipids (or lipid cofactors), which are tightly bound in crevices or between subunits of the protein and are often crutial for the activity of the protein. Electron paramagnetic resonance (EPR) can be used to measure the mobility of annular lipids, and thus gives information on the selectivity for annular lipids of a protein. Most proteins are found to prefer negatively charged lipids such as phosphatidylserine and phosphatidylinositol.
Strong lipid-protein interactions are also evident form crystal structures, and this interaction can be specific. For example, three cardiolipin (CDL) molecules were found to bind to three sites on the ANT1 transporter. The tendency of CDL to bind to these sites can also be shown in molecular dynamics simulations. The specificity of this binding can also be shown computationally, by calculating the free energy at different protein-lipid separations for CDL and other lipids (Figure 6.1).
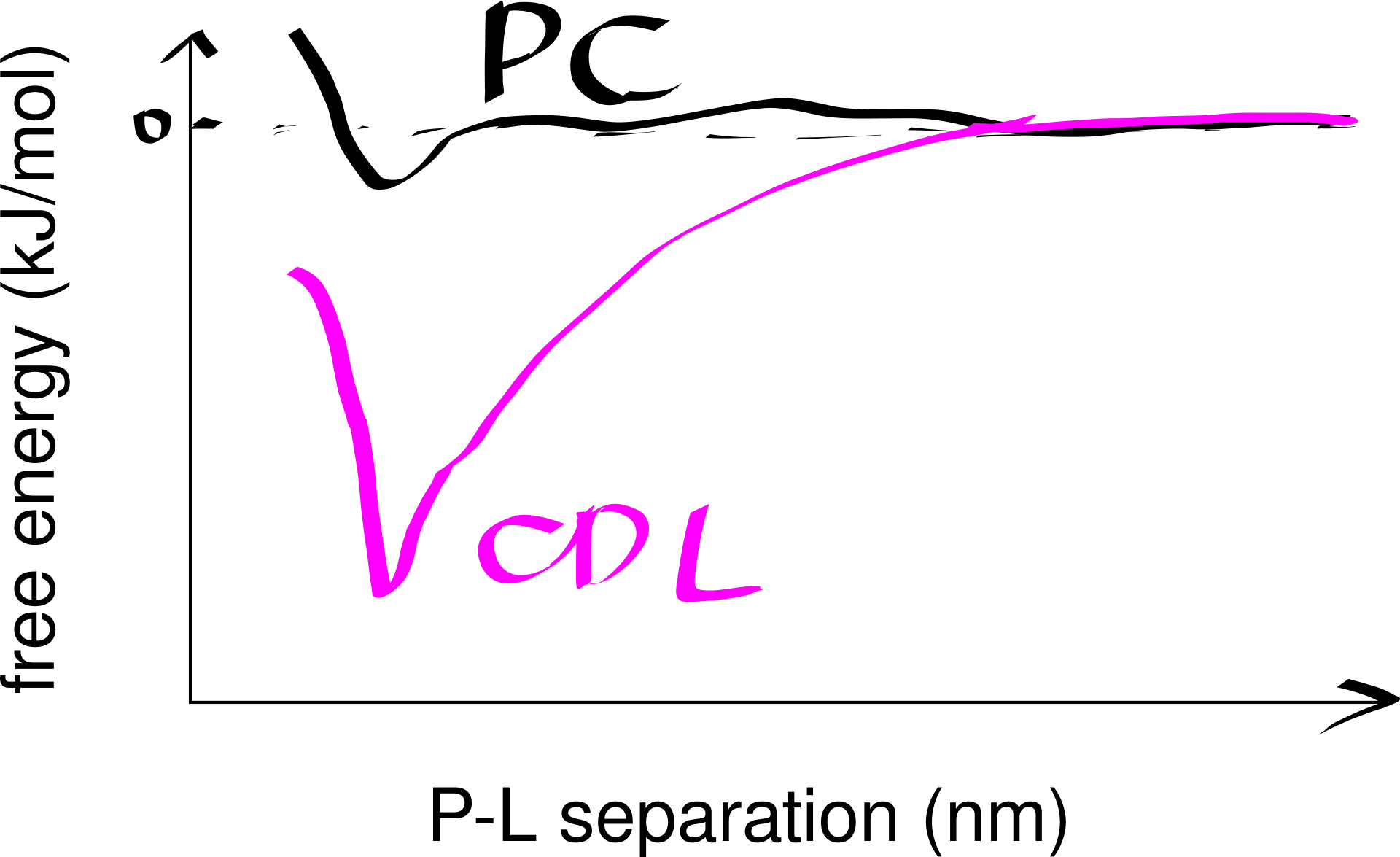
Figure 6.1: Computational analysis of a protein’s preference to bound lipids. CDL binding results in a significantly lower energy state.
Inward-rectifier K+ channels (Kir) are an example where the lipid-protein interaction is directly involved in the protein’s function. Kir have a Transmembrane domain (TMD) and an intracellular C-terminal domain (CTD). PIP2 binding cuases the CTD to move towards the TMD, which in turn causes the helices in the TMD to bend and thus opens the gate (Figure 6.2).
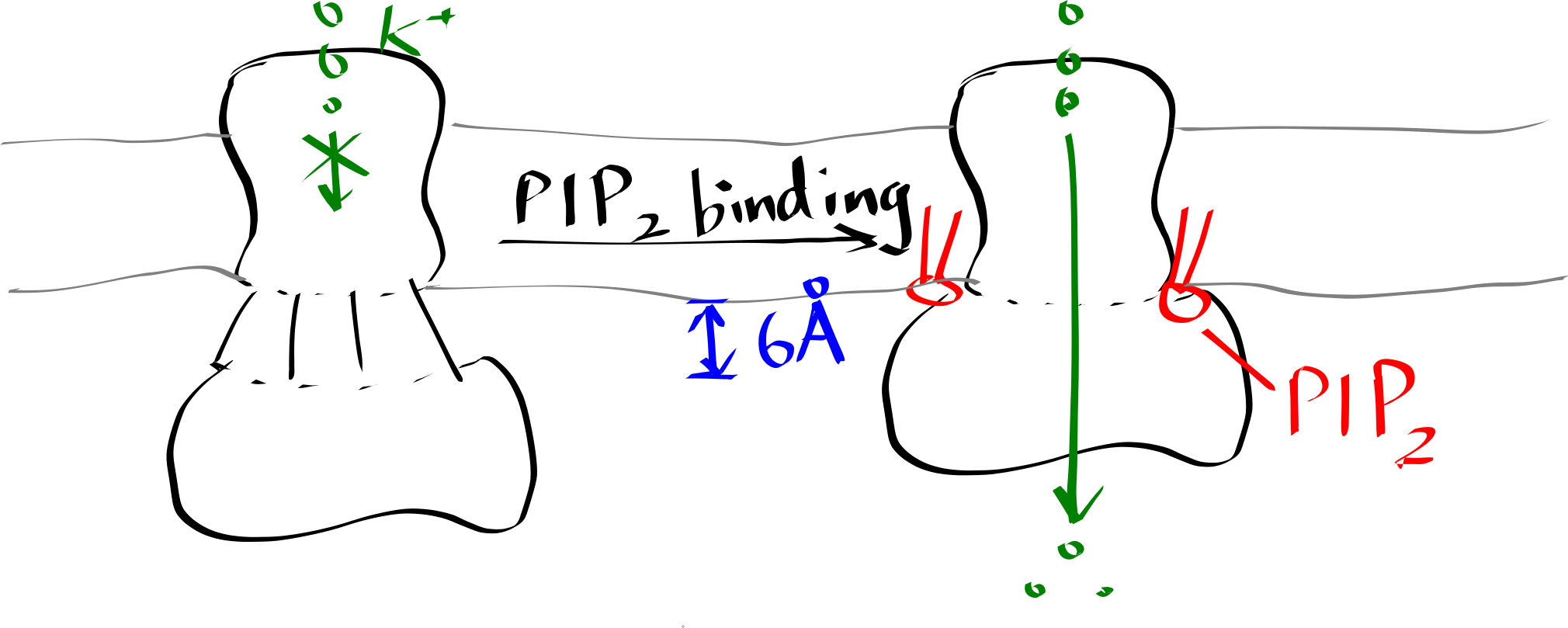
Figure 6.2: Lipid-gated Kir channel
Many crystal structures of GPCRs reveal cholesterol bound to the protein. Molecular dynamics simulations at different cholesterol concentrations suggests the role of cholesterol in reducing the dynamic flexibility the protein and stabilising a certain conformational state in an allosteric fashion.